 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipRemoving palladium has become more and more difficult for a number of reasons. Industry is using it more frequently, due to its greener credentials as a catalyst but regulatory bodies are also reducing acceptable limits in APIs. One of the most significant changes for pharma recently has been increased scrutiny of impurities in pharma products.
These changes have been in the making for the past 15 years, edging closer and closer to formal policy in recent years by various regulatory bodies. In 2016 the US Food and Drug Administration (FDA) issued guidelines for the analysis of elemental impurities in drugs, drug products (as defined in ICH Q6A and Q6B), raw drug materials and dietary supplements, harmonizing USP 232 & 233 with the requirements of ICH Q3D. The European Medicines Agency (EMA) had already adopted ICH Q3D guidance for new marketing authorisations, extended to existing authorised medicinal products from December 1st 2017. All manufacturers of drug products marketing products in the USA and Europe essentially now need fully comply with the requirements of ICH Q3D.
Global Standards of Impurity Control
The ICH (International Conference Harmonization) Q3D Guideline for elemental impurities presents major challenges to testing and risk assessment within the pharmaceutical industry. Q3D considers the type of metal impurity in a drug product, its mode of action along with scientific and risk-based approaches to control. We’ve put together a how-to guide for this important topic.
How do I comply?
- Identify known and potential sources of elemental impurities for the drug product.
- Evaluate the presence of an elemental impurity in the drug product by determining the observed or predicted level of the impurity and comparing with the established PDE.
- Summarize and document the risk assessment. Identify if controls built into the process are sufficient or identify additional controls to be considered to limit elemental impurities in the drug product.
What are the implications for my process or synthesis?
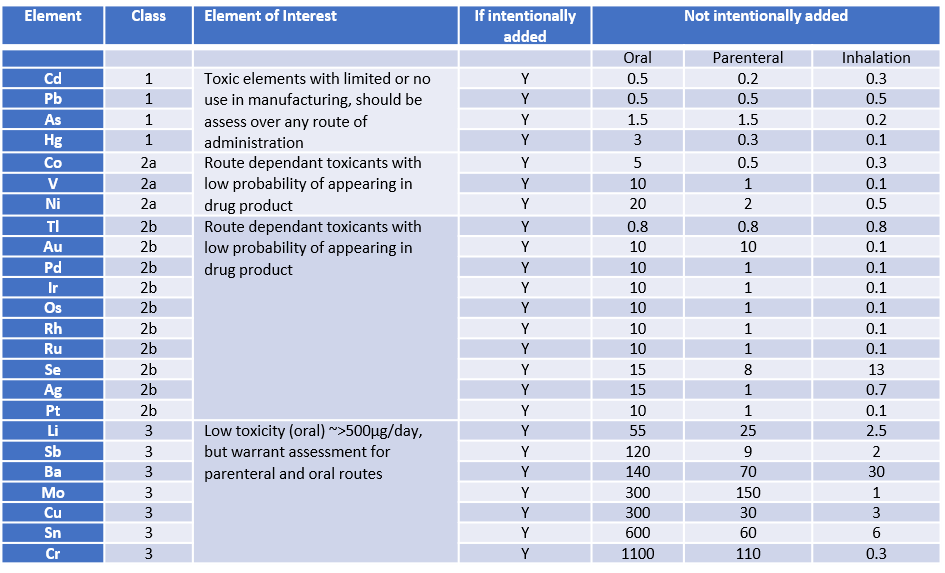
Its pretty simple at face value, if any key metals (see table below) are intentionally added during a synthesis or delivered to you in the form of a raw material from one of your suppliers, they must be risk assessed. Regardless of that, all class 1 and 2a metals must be screened and risk assessed. And in general class 3 metals needs only be assessed if the final route of administration is inhalation or injection. Lower-risk elements (Fe, B, W, Zn, K, Ca, Na, Zr, Mn, and Mg) were not considered by the Q3D Expert Working Group (EWG) to require route specific PDEs and do not need to be included in a risk assessment, unless required by regional or local regulations.
ICH Q3D comprises of 3 parts. Whilst the rules are complex, the process can be broken down into 3 steps.
- Evaluation of the toxicity data for potential elemental impurities
- Establishing a Permitted Daily Exposure (PDE) for each element of toxicological concern
- Conduct a risk-based approach to control elemental impurities in drug products
ICH Q3D covers 24 elements (classified under the following classes 1, 2A, 2B and 3) and gives permitted daily exposure (PDE) in drug products:
What about ppm? – isn’t that important?
Yes, it is, but pharma PDEs are reported in micrograms of the drug product per day. Since ppm is easier to visualize as metric, PDE levels are often converted into ppm. Metals PPM is often a better metric of the risk assessment and often leveraged as a control of quality in a chemical process.
There are 3 options for calculating ppm
- Option 1: Calculated concentration (µg/g (ppm) = PDE (µg/day)/daily amount of drug product (g/day)
- Option 2: Accept either common limits across ingredient in drug products or (2b) allow individual limits for elements in the upstream processing of a drug product.
- Option 3: measure the final drug product directly (may be too late if a large process change is needed)
Based on this, and general principles of risk avoidance, good practice employs metal mitigation strategies where necessary during a chemical synthesis thus reducing the chance of downstream contamination issues.
OK, so what are the practical solutions for compliance?
Traditional methods to remove metals include chromatography, activated carbon, extraction, distillation and recrystallization, but these can offer poor selectivity and lead to high API loss. In some cases, crystallization can actually concentrate a metal within a crystal structure. The use of metal scavengers has significantly increased in popularity due to their ability to be added into a process in a mechanically simple way, they are compatible with existing process equipment, and easily removed from reactions afterwards.
Since there are potentially a lot of metals to be removed, and a lot of scavengers, the main issue is one of choice. We’ve seen very large studies by well-funded (both time and money) pharma groups employing a lot of screening and scavengers but thorough analysis takes time, one must find the right scavenger and then find the right conditions for its use, and that may vary depending on the pharma product itself!
That’s why we have put together a guide and developed robust metal scavenging SOPs to significantly simplify this process. Jump to the next article on screening approaches to learn more.